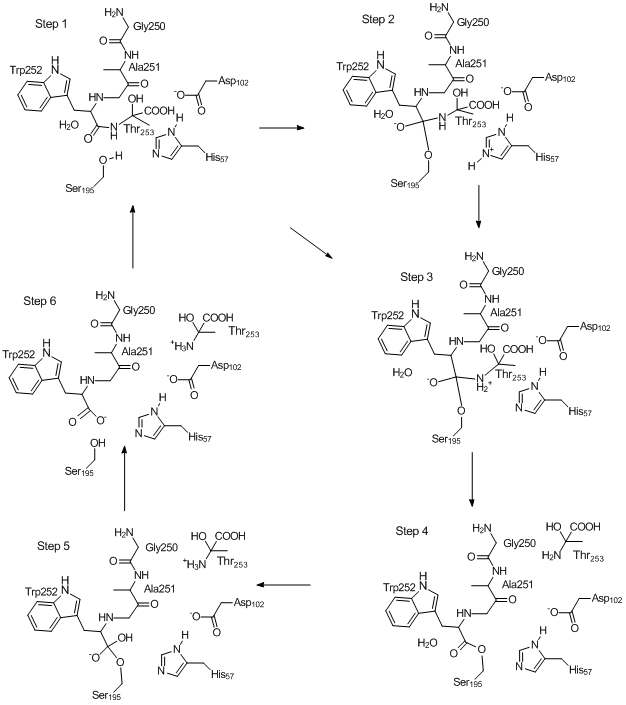
Mechanism of Chymotrypsin Catalyzed Hydrolysis of Peptide Bond
(Back to "Proteins")
The mechanism whereby chymotrypsin catalyzes the hydrolysis of a peptide bond
can be represented in several steps as shown in the figure below. Each step can be defined as a
stationary point on the Potential Energy Surface (PES). These stationary points
were calculated using PM7, and can be investigated in detail in the links named
"Step n" below. The starting point was 8GCH.pdb.
In 8GCH the substrate is the tripeptide Gly250-Ala251-Trp252, with Trp252
terminated by a carboxylic acid group. This sequence also occurs in
chymotrypsin at residues 205, 206, and 207, with residue 208 being Thr, so it is
reasonable to assume that the substrate is the product of hydrolysis of the
Trp-Thr peptide bond in a chymotrypsin by the chymotrypsin in 8GCH. In
order to begin modeling the chymotrypsin mechanism, one of the oxygen atoms on carboxylate on Trp252 was replaced by Thr253,
this giving a peptide bond ideally positioned for the simulation described here.
All the steps
have the same formula, C1115H2450N301O706S16,
or 4,588 atoms, so a comparison of the various structures can be made. To
reproduce the working shown on this page and those linked to it, download the
ARC files for Steps 1 to 6 (on this page) and follow the instructions for
locating and
refining transition states in proteins.
NOTE: After this work was published, a method for
calculating the interaction energy of a ligand non-covalently bound to a protein
was developed. This required increasing the accuracy of the geometry
optimizations, and resulted in several changes being made to MOPAC.
Because of this, there will be significant differences in the various heats of
formation cited here, and much smaller differences in derived quantities such as
relative energies.
Step 1: Substrate is
docked in active site. This geometry can be arbitrarily defined as the
starting point of the catalytic mechanism. (ARC
file)
Step 2: The Ser195 hydroxyl oxygen bonds to
the peptide carbon, and its proton migrates to His57. (carbon, and its proton migrates to His57. (ARC file)
Step 3: The
proton on His57 migrates to the nitrogen atom of the peptide bond being
hydrolyzed, resulting the the formation of a Zwitterion.
The anionic oxygen attached to the tetrahedral carbon is stabilized by
hydrogen bonding to the oxyanion hole. (ARC file)
Step
4: The now-unstable Zwitterionic peptide bond breaks, forming an
ester and a chain fragment (here Thr253)
which then migrates out of the active site. (ARC
file)
Step
5: A water molecule adds to the ester to re-form a tetrahedral
carbon. The departing Thr253 ionizes. (ARC file)
Step 6: The proton on
the tetrahedral carbon migrates to Ser195, splitting the Ser195 - Trp252 ester bond
to form an acid plus alcohol. (ARC file)
Notes: Each step
represents a different geometry, i.e., a set of coordinates. The
difference between each set of coordinates can be expressed as the sum
of the differences in positions of equivalent atoms in the two sets of
coordinates. Thus for
"Distance" between different
Stationary Points (Ångstroms)
Step 1
Step 2
Step 3
Step 4
Step 5
Step 6
Step 1
0.0
Step 2
164.1
0.0
Step 3
80.1
116.3
0.0
Step 4
118.1
115.4
59.1
0.0
Step 5
215.1
208.0
188.5
192.2
0.0
Step 6
273.9
228.9
168.1
0.0
Step 1 and Step 2, the main difference is the formation of the
tetrahedral carbon involving Ser195 covalently bonding to Trp252, and serine's hydroxyl hydrogen migrating to the imidazole ring of
His57. This movement is quite small, but when the motions
of all the atoms are included, the result is very large.
The motion in going from one step to the next step looks large, but
if even a small bias pulling one geometry in the direction of another is
applied, then the "distance" between the geometries would drop considerably.
For six steps, there are 15 = (6·5)/2 possible transition
state. Of these, five are "real" in the sense that they represent
barriers between adjacent steps, the other ten transition
Heats of Formation of
Steps and Transition States (kcal/mol)
states do not represent anything meaningful in a chemical sense.
The transition state between Step 3 and Step 4 is activationless - the
Thr353 cation spontaneously migrates out of the active site.
The
transition state between Step 2 and Step 4 collapses back to the
transition state between Step 1 and Step 2, indicating that this is a
nonsense reaction.
The lowest energy pathway is Step 1 →
Step 2 → Step 3 → Step 4 → Step 5 → Step 6, i.e.,
the classical chymotrypsin mechanism.
Energy differences are precise to ~±2 kcal/mol, and are of unknown
accuracy. The loss of precision is due to the very flat PES at the local energy
minimum. The heats of formation given here represent the fully optimized
systems, i.e., stationary points on the PES. Even small changes, such as
proton migration from Step 2 to Step 3 result in a a large increase in energy,
typically several tens of Kcal/mol, and only after the system relaxes completely are
the ΔHf given here obtained.