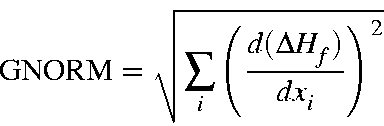
In normal (non-publication quality) work the default precision of MOPAC is recommended. This will allow reasonably precise results to be obtained in a reasonable time. Unless this precision proves unsatisfactory, use this default for all routine work.
The best way of controlling the precision of the geometry optimization and gradient minimization is by specifying a gradient norm which must be satisfied. The gradient norm is the scalar of the vector of derivatives of the energy with respect to the geometric variables flagged for optimization. I.e.,
where xi refers to coordinates flagged for optimization. Note that the calculated GNORM may be very small and at the same time the geometry might not be at a stationary point. This can easily happen when (a) less than 3N-6 coordinates are flagged for optimization, (b) SYMMETRY has been used incorrectly, or (c) (less common) only 3N-6 coordinates are flagged for optimization and dummy atoms are used. If any one of these three conditions occurs, then the warning message, "WARNING - GEOMETRY IS NOT AT A STATIONARY POINT" will be printed.
A less common, but not unknown, situation arises when internal coordinates are used. In this strange situation, the internal derivatives might all be zero, but the Cartesian derivatives are large. An example of such a system is shown in the Figure.
Figure:
Example of Spurious Stationary Point|
Line 1: GRADIENTS 1SCF DEBUG DCART Line 2: EXAMPLE OF INTERNAL COORDINATE DERIVATIVES ZERO Line 3: AND CARTESIAN DERIVATIVES LARGE Line 4: O Line 5: C 1.2075664 1 Line 6: H 1.2114325 1 103.347931 1 0 0 2 1 Line 7: H 1.0904182 1 180.000000 1 90 1 2 1 3 |
In this strange system, the bond-angle of the second hydrogen is 180°, and the dihedral is 0°. Obviously, the derivative of the dihedral will be zero. The derivative of the angle is not so obvious. If the angle changes, then the fourth atom will move out of the plane of the other three atoms. The energy will change in the same way regardless of whether the angle increases or decreases; therefore, the derivative of the angle must be zero.
Because of the unusual nature of this type of system, users may be unaware of the danger. If such a system is detected, a warning will be given and the job stopped. For all other cases, the "WARNING - GEOMETRY IS NOT AT A STATIONARY POINT" message will be printed on completion of the calculation. In the unlikely event that the calculation should not be stopped when the strange system is detected, calculation can be continued by specifying GEO-OK.
Modification of GNORM is done via the keyword GNORM=n.nn. Altering the GNORM automatically disables the other termination tests resulting in the gradient norm dominating the calculation. This works both ways: GNORM=20 will give a very crude optimization while GNORM=0.01 will give a very precise optimization. The default is GNORM=1.0.
When the highest precision is needed, such as in exacting geometry work, or when you want results whose precision cannot be improved, then use the combination keywords GNORM=0.0 and either RELSCF=0.01 or SCFCRT=1.D-NN; (NN should be in the range 5-15). By default, EigenFollowing is used in geometry optimization. One reason is that EigenFollowing is nearer to a gradient minimizer than it is to an energy minimizer. Because of this, if there is any difference between the gradient minimum and the energy minimum, it will give better reproducibility of the optimized geometry than the alternative BFGS method.
In practice, optimized geometries for "well behaved" systems can be obtained with GNORMs of less than 0.0001.
Increasing the SCF criterion (the default is SCFCRT=1.D-4) improves the precision of the gradients; however, it can lead to excessive run times, so take care. Also, there is an increased chance of not achieving an SCF when the SCF criterion is excessively increased.
Superficially, requesting GNORM=0 might seem excessively stringent, but as soon as the run starts, it will be cut back to 0.01. Even that might seem too stringent. The geometry optimization will continue to lower the energy, and hopefully the GNORM, but frequently it will not prove possible to lower the GNORM to 0.01. If, after 10 cycles, the energy does not drop then the job will be stopped. At this point you have the best geometry that MOPAC, in its current form, can give.
If a slightly less than highest precision is needed, such as for normal publication quality work, set the GNORM to the limit wanted. For example, for a flexible system, a GNORM of 0.1 to 0.5 will normally be good enough for all but the most demanding work.
If higher than the default, but still not very high precision is wanted, then use the keyword PRECISE. This will tighten up various criteria so that higher-than-routine precision will be given.
If high precision is used, so that the printed GNORM is 0.000, and the resulting geometry resubmitted for one SCF and gradients calculation, then normally a GNORM higher than 0.000 will result. This is not an error in MOPAC: the geometry printed is only precise to eight figures after the decimal point. Geometries may need to be specified to more than eight decimals in order to drive the GNORM to less than 0.000.
If you want to test MOPAC, or use it for teaching purposes, the GNORM lower limit of 0.01 can be overridden by specifying LET, in which case you can specify any limit for GNORM. However, if it is too low the job may finish due to an irreducible minimum in the heat of formation being encountered. If this happens, the "STATIONARY POINT" message will be printed.
Examples of highly optimized geometries can be found in the port.dat file. When this job is run, most gradients will be less than 0.001 kcal/mol/Å. A few will be larger. These exceptions fall into two classes: diatomics, for which a simple line-search is sufficient to locate the optimum geometry, in which case the GNORM criterion is not used; and non-variationally optimized systems, where the analytical C.I. derivatives are used. These derivatives are of lower precision than the variational derivatives, but are still much better than finite difference derivatives using full SCFs. Finally there is a full analytical derivative function [16] within MOPAC. These use STO-6G Gaussian wavefunctions because the derivatives of the overlap integral are easier to calculate in Gaussians than in STOs. Consequently, the
Some calculations, mainly open shell RHF or closed shell RHF with C.I., have untracked errors which prevent very high precision. For these systems GNORM should be in the range 1.0 to 0.1.