Sometimes in a MOZYME calculation, a Lewis structure cannot be constructed from
the topology of the system supplied. When that happens, a warning message will be printed and
the run stopped. Unwanted connections can be removed using CVB.
Two formats are supported:
Using atom numbers (if possible, use
atom labels for proteins)
CVB(n1:-n2[;n3:-n4[;n5:-n6....]]])
where the connections to be removed are those between atoms n1-n2, n3-n4, n5-n6, etc.
See atom numbering. Thus if
connections were to be removed between atoms 4 and 5, and between atoms 665 and 670, then
CVB(4:-5;665:-670) would be used. If connections are needed, then don't use the minus sign. Thus if
connections were
needed between atoms 4 and 5, and between atoms 665 and 670, then
CVB(4:5,665:670) would be used. CVB is also useful when
RESIDUES and PDBOUT
are present. A topology sometimes has an
unwanted connection between two chains that prevents some of the residues being
identified. To correct this fault, remove the unwanted connection using CVB.
If there are a several unwanted connections , and these connections involve a
specific element, then consider the alternative of using VDWM to change the Van der Waals radius for that
element.
Here Labeln is the label, in PDB or JSmol format, for the atom.
Using the PDB format, the label starts
with the first non-blank character of the atom name and ends with the last
character of the residue number. For example: CVB("2HB SER B
4":-"OD1 ASP B 119","1HH1 ARG B 5":"C TYR B 7").
Spaces are not important, but using spaces makes it easier to read the atom label.
With JSmol, the label starts with a "[" and ends at the character before " #".
As with PDB format, spaces are not important when JSmol format is used, i.e., CVB("[LYS] 4 : A . CD":"[LYS]4:A.CG"). The advantage of
using atom labels
is that if atoms are added or removed the atom label would not change.
Another advantage is that PDB TER lines, positional disorder, and other things
that cause the PDB line number to not match the atom number do not cause
problems.
Wildcards are allowed for the residue name and for the chain letter.
When a wildcard is used, a letter is replaced by an asterisk, "*", thus:
CVB("2HB *** B 4":-"OD1 *** * 119","1HH1 ARG * 5":"C TYR B 7") and CVB("[***] 4 :
* . CD":"[***]4:*.CG"). The first occurrence that matches
will be used.
Special case of O-O bonds.
When proteins that contain a large number of water molecules are edited,
quite often one or more water molecules become badly positioned, resulting in
spurious O-O interactions. These can be eliminated using the standard
options within the CVB keyword, but as this problem occurs quite often, a
special option, "O:-O" is provided. This six-character option will
identify and delete all spurious O-O bonds.The option can be anywhere between
the opening "(" and closing ")" of the CVB keyword.
Intermediates in Enzyme Mechanisms
A frequent problem when modeling intermediates and transition states in
enzyme mechanisms is that the structure cannot be written in Lewis form. An
example occurs in the transition state when a cystine sulfur starts to bond to
the carbon of a guanidinium group in an Arg, and the hydrogen that was on the
sulfur migrates to one of the nitrogen atoms of the guanidinium. If this
happens, the
resulting -NH3 group would have a positive charge and the carbon
would be given a negative charge. (A carbon atom bonded to a cation and having only three
valencies used would automatically be assigned a negative charge.) Sulfur having
only one bond (to its CH2 group) is, by default, also given a negative charge.
The net charge on this assembly is now -1 instead of the expected +1. To
correct this error CVB is used to make a bond between S and the H of -NH3.
This automatically causes the N-H bond to be deleted giving -NH2, a
component of the
normal guanidinium group. Now that sulfur is bonded to its hydrogen as
will as to its CH2 group, and
the guanidinium has the usual +1 charge, the net charge in this region is
correct.
A guiding principle would be to use CVB to make the structure as similar to either the
reactant or product as possible.
Note that CVB is used only in the generation of a starting Lewis
structure or in generating a PDB file. The presence of unrealistic connections in this structure will not
normally give rise to an incorrect SCF.
See also SETPI to explicitly assign π bonds, and
atom labels to explicitly assign charges.
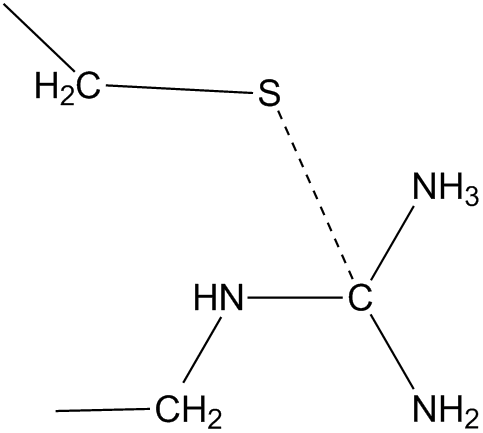 A frequent problem when modeling intermediates and transition states in
enzyme mechanisms is that the structure cannot be written in Lewis form. An
example occurs in the transition state when a cystine sulfur starts to bond to
the carbon of a guanidinium group in an Arg, and the hydrogen that was on the
sulfur migrates to one of the nitrogen atoms of the guanidinium. If this
happens, the
resulting -NH3 group would have a positive charge and the carbon
would be given a negative charge. (A carbon atom bonded to a cation and having only three
valencies used would automatically be assigned a negative charge.) Sulfur having
only one bond (to its CH2 group) is, by default, also given a negative charge.
The net charge on this assembly is now -1 instead of the expected +1. To
correct this error CVB is used to make a bond between S and the H of -NH3.
This automatically causes the N-H bond to be deleted giving -NH2, a
component of the
normal guanidinium group. Now that sulfur is bonded to its hydrogen as
will as to its CH2 group, and
the guanidinium has the usual +1 charge, the net charge in this region is
correct.
A frequent problem when modeling intermediates and transition states in
enzyme mechanisms is that the structure cannot be written in Lewis form. An
example occurs in the transition state when a cystine sulfur starts to bond to
the carbon of a guanidinium group in an Arg, and the hydrogen that was on the
sulfur migrates to one of the nitrogen atoms of the guanidinium. If this
happens, the
resulting -NH3 group would have a positive charge and the carbon
would be given a negative charge. (A carbon atom bonded to a cation and having only three
valencies used would automatically be assigned a negative charge.) Sulfur having
only one bond (to its CH2 group) is, by default, also given a negative charge.
The net charge on this assembly is now -1 instead of the expected +1. To
correct this error CVB is used to make a bond between S and the H of -NH3.
This automatically causes the N-H bond to be deleted giving -NH2, a
component of the
normal guanidinium group. Now that sulfur is bonded to its hydrogen as
will as to its CH2 group, and
the guanidinium has the usual +1 charge, the net charge in this region is
correct.