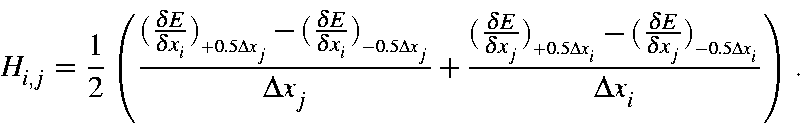The Hessian matrix is the matrix of second derivatives of the energy with respect to geometry. The most important Hessian is that used in the FORCE calculation. Normal modes are expressed as Cartesian displacements, consequently the Hessian is based on Cartesian rather than internal coordinates. See also Vibrational Relationships - Derivation.
Although first derivatives are relatively easy to calculate, second derivatives are not. The simplest, although not an elegant, way to calculate [73] second derivatives is to calculate first derivatives for a given geometry, then perturb the geometry, do an SCF calculation on the new geometry, and re-calculate the derivatives. The second derivatives can then be calculated from the difference of the two first derivatives divided by the step size. This method, which is used in the EigenFollowing routine, is called "single-sided" derivatives.
The Hessian is quite sensitive to geometry, and should only be evaluated at stationary points. Because of this sensitivity, "double-sided" derivatives are used.
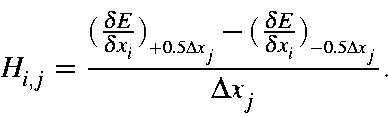
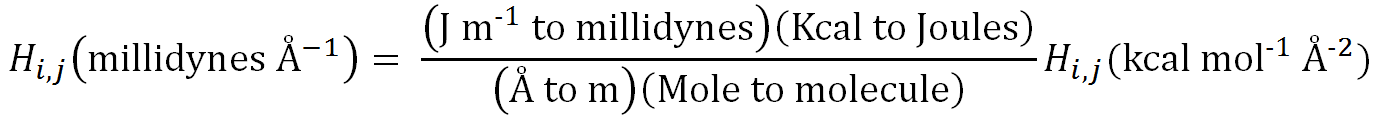
The conversion from (J m-1) to millidynes is also unusual in that
it involves two steps. Expanded, this involves converting (J m-1)
to dynes, followed by converting dynes to millidynes. This involves the
factors 105 times 103, so the total conversion factor is
108,
as shown in the following expression:
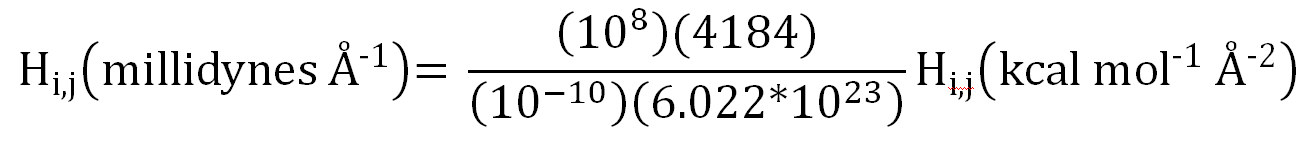
This matrix represents the force constants for the system. In order
to calculate the vibrational frequencies it must first be
mass-weighted:
In this expression, Mi and Mj are the atomic weights in amu, so for a nitrogen molecule Mi = Mj = 14.0067.
Diagonalization of this matrix yields eigenvalues, εi, from which the vibrational frequencies, νi can be calculated. For simple harmonic motion involving a mass, m, and force-constant, k, the frequency, f, in cycles per second is given by:
f = 1/(2π) x (k/m)½ cycles per second
But, by convention, vibrational frequencies in UV-Visible spectroscopy are reported in reciprocal centimeters. To convert from f to ν, f must be divided by the speed of light, c.
| ν | = f/c |
| = 1/(2πc) x (k/m)½ | |
| = 1/(2πc) x (εi)½ cm-1 | |
At this point, some quantities are not in the correct units. The eigenvalues are in millidynes per Ångstrom
per amu. Before they can be used they must be converted into SI units,
by using:
1 millidyne per Ångstrom = 102 J m-2,
Atomic
mass units (amu) = 10-3 x N-1 kilograms.
Combining these two factors together:
Millidynes per Angstrom per amu = (102 x 103 x N) J m-2 kg-1 = (105 x N) Newton m-1 kg-1
Once this is done, the frequency in cm-1 can be calculated:
| ν | = 1/(2πc)(105 x N x ε)½ |
| = 1/(2 x 3.1416 x 2.9979 x 1010)) x (6.022 x 1028 x ε)½ | |
| = 1302.79 x ε½ cm-1 |
The eigenvalue also represents the quantity (k/μ)1/2 , where k is the force constant and μ is the reduced mass, but further discussion of this will be left until later.