GEO_REF supplies a second geometry to a job. If
more than one line of keywords is supplied, then this keyword should be on the
first line.
There are two ways to specify "text" :
(A) Specify the file name and location, e.g.:
GEO_REF="CRAMBIN 1CBN X-ray.mop"
GEO_REF="CRAMBIN.arc"
GEO_REF="CRAMBIN.pdb"
GEO_REF="./CRAMBIN.arc"
GEO_REF="../CRAMBIN.arc"
GEO_REF="../../arc files/CRAMBIN.arc"
GEO_REF="sub folder/CRAMBIN.arc"
GEO_REF="./sub
folder/CRAMBIN.arc"
GEO_REF="M:\data sets\CRAMBIN 1CBN X-ray.mop"
GEO_REF="/Users/jstewart/data_sets/CRAMBIN_1CBN_X-ray.mop"
GEO_REF="~/data_sets/CRAMBIN_1CBN_X-ray.mop"
(B) Use
GEO_REF="SELF". When "SELF"
is present, a file that has the same name as the MOPAC data-set can be used.
By default, this data-set would be the data-set in the current job. An
alternative would be to use a data-set in a different folder, for example:
GEO_REF="../Reference geometry/SELF"
GEO_REF="../../arc files/SELF"
GEO_REF="C:/PM6 geometries/Self"
If desired, the suffix can be changed, so if the data-set suffix is
".mop" and the file that is to be used as the
GEO_REF file has the suffix ".arc", then replace "SELF"
with "SELF.arc"
If GEO_REF="SELF" is used, then the GEO_REF geometry used will be the one defined in the
MOPAC data-set, even if RESTART is also present. That is, the
GEO_REF geometry used will be the geometry before any changes caused by RESTART
are applied. This allows large systems to be restarted easily.
GEO_REF can be used for
comparing geometries, improving X-ray geometries and
also for locating
transition states (using LOCATE-TS or
SADDLE).
In all cases, the job uses two geometries, one from the data set or defined
using GEO_DAT, and one from a
second data set defined by the keyword
GEO_REF. By default, the GEO_REF system will be rotated and
translated to minimize the difference between it and the GEO_DAT
geometry. This operation will not be carried out if keyword NOREOR
is present. This allows a strict comparison of closely-related systems in
which some atoms are frozen.
If the file containing the geometry also contains keywords, job name, etc. these
will be ignored. Only the keywords, job name, etc., in the MOPAC data set,
i.e., the file containing
GEO_REF=<text>, will be used. Some keywords, such as
CVB, may also require GEO-OK.
Both systems will be converted to simple Cartesian
coordinates.
Any dummy atoms supplied in either the first system or the system defined by
GEO_REF will be ignored, as will any translation vectors.
Requirements
MOPAC data sets must contain PDB information. Before any calculation is started, the two data sets are examined and
compared. For both PDB files and for MOPAC data sets, the examination consists of checking that every atom has a
unique label. If any of the following strict requirements is not
met,
an error message printed and the job will be stopped:
- Both species must have the same empirical formula
- For PDB files and for MOPAC files with PDB information, all atom labels must be unique. Atom numbers don't count towards the
label.
-
For PDB files and for MOPAC files with PDB information, the
same labels must be present in both the reference and job data sets.
It is normal for this test to fail the first time it is run. Use the
error message to identify the fault, then correct the fault and re-run the job.
When no errors are reported, the data set labels are acceptable.
The following flexibilities are allowed:
- If the only operation is comparing two geometries, then the species can
have different empirical formulae. Also, some atoms can have different
labels, but all such atoms will be excluded from the comparison.
- The order of atoms does not need to be the same. If the order is
different, the reference data set will be re-ordered to match the job data
set.
- Where there is ambiguity, e.g., Cδ1
and Cδ2 in Phe and Tyr. Ambiguities of this
type are automatically resolved by re-labeling atoms in the reference data
set, unless NOSWAP is
present.
From a practical point of view, this flexibility means that the order of
atoms in the two data sets is not important.
The reference data set will then be rotated and translated to give the best
overlap with the job data set. This new geometry will be printed as
<filename>.new Total geometric differences between the two structures
will be printed in the output.
Comparing geometries
If the keywords GEO_REF,
0SCF
and HTML are all present, or if
COMPARE is present, then an HTML web-page
will be written that allows pairs of structures to be compared.
If there are some atoms in one data-set that are not in the other, or vice
versa, then the job will be stopped. However, if
GEO-OK is present then the job will continue, but using only the atoms that are
common to both data-sets.
Improving X-ray geometries
The accuracy of native structures, X-ray and NMR, can be increased by
performing a limited geometry optimization, starting with the conditioned native
structure.
The optimized geometry is a hybrid of the PM7 geometry, assuming that PM7 is
used, and the native geometry. The hybrid heat of formation, H', is
given as:
H' =ΔHf(PM7) + cΣ(Xi
- Xi(0))2
That is, at each geometry, a restraining potential is added
to the PM7 heat of formation.
To specify a restraint, put the new value after the
reference data set, e.g., if a value of 10 is wanted, use:
GEO_REF="M:\data_sets\CRAMBIN_1CBN_X-ray.mop"10
or
GEO_REF="CRAMBIN_1CBN_X-ray.mop"10.
The effect of the restraining potential is to reduce PM7 errors in the
secondary, tertiary, and quaternary structures. It has very little effect
on the primary structure, e.g., bond lengths and angles.
The effect of the restraining potential can be illustrated using Crambin,
1CBN. In the original structure there was an extra C2O moiety, this was
removed. 1CBN has both structural and positional disorder; the sequence
used was:
TTCCPSIVARSNFNVCRLPGTPEALCATYTGCIIIPGATCPGDYAN This sequence
differs from the later default sequence of TTCCPSIVARSNFNVCRLPGTSEALCATYTGCIIIPGATCPGDYAN
(difference in bold) The formula used was C202H315N55O64S6,
i.e. 642 atoms. Because the positions of the hydrogen atoms were severely
incorrect in the X-ray structure, before starting any comparison of the effect
of GEO_REF on the geometry, their positions were optimized.
During geometry optimization, the COSMO solvation model was used because some
residues in Crambin were reported as being ionized.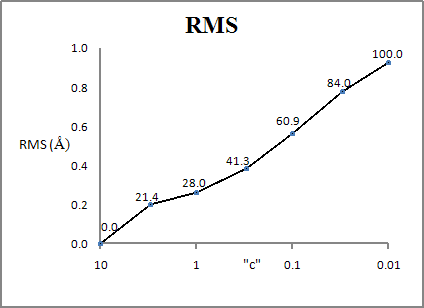
 The difference in heat of formation of
Crambin, after
preconditioning (that is, after the positions of the hydrogen atoms were
optimized), and after
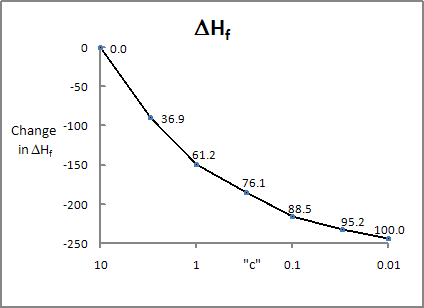
complete geometry optimization, is ~250 kcal mol-1. The RMS change in
geometry is about 1.0 Ångstroms. If the
geometry is optimized using GEO_REF and a constant, c, of 1.0, then the heat of formation drops by
~150 kcal mol-1, and the RMS geometry change is ~0.25
Ångstroms. In other words, by allowing the X-ray geometry to change by 0.25
Ångstroms, over half of the strain energy in the X-ray structure is removed.
Put yet another way, the resulting geometry is more than twice as accurate, in terms of
chemistry, than the X-ray structure. All
Crambin structures used here.
The difference in heat of formation of
Crambin, after
preconditioning (that is, after the positions of the hydrogen atoms were
optimized), and after
complete geometry optimization, is ~250 kcal mol-1. The RMS change in
geometry is about 1.0 Ångstroms. If the
geometry is optimized using GEO_REF and a constant, c, of 1.0, then the heat of formation drops by
~150 kcal mol-1, and the RMS geometry change is ~0.25
Ångstroms. In other words, by allowing the X-ray geometry to change by 0.25
Ångstroms, over half of the strain energy in the X-ray structure is removed.
Put yet another way, the resulting geometry is more than twice as accurate, in terms of
chemistry, than the X-ray structure. All
Crambin structures used here.
Locating transition states
GEO_REF can also be used for moving a reactant or product geometry in the
direction of the transition state. Consider two data sets, reactant.mop
and product.mop in folder M:\, in which the heat of formation of the optimized
product geometry is lower than that of the optimized reactant geometry.
The product geometry geometry can be moved in the direction of the transition
state by using keyword
GEO_REF="M:\reactant.mop"
The .arc file can then be edited to give a new data set, product_on_slope.mop.
The reactant geometry can then be moved in the direction of this new geometry by
using keyword
GEO_REF="M:\product_on_slope.mop"
Again, edit the .arc file to give reactant_on_slope.mop. Why was the
product moved first? Because by moving it towards the reactant geometry,
its heat of formation would rise in proportion to the distance to the reactant
geometry. When the reactant geometry is moved towards the product geometry on
the slope, the distance from the starting reactant geometry to the product
geometry on the slope is less, so the rise in energy would be less.
If keyword TS is present in a GEO_REF calculation, the optimization is not
run. Instead, the two geometries are averaged, and the result written to a
new file <file>.new.
For the current exercise, a good approximation to the transition state can
then be generated from the data set reactant_on_slope.mop by using keywords TS
and
GEO_REF="M:\product_on_slope.mop"
Error: "Fault detected in atom labels in a GEO_REF run"
If the only faults detected are those that involve hydrogen atoms, then add
keyword GEO-OK, and do not have NOSWAP,
and re-run. The hydrogen atoms will be put in their correct place
automatically. Alternatively, edit either
the data-set or the GEO_REF file to remove the fault.