Example of how to handle Symmetry Disorder in a CSD file
EMETAS (all files)
Disorder in molecule orientation and in molecule position is a natural phenomenon in crystal structures. A good example of these phenomena is provided by CSD entry EMETAS, hexa-aqua-magnesium(ii) bis(2-(2-pyridyl)pyridinium) bis(adenosine-5'-triphosphate)-magnesium(ii) hexahydrate, where all the bis(2-(2-pyridyl)pyridinium) or protonated 2,2'-dipyridylamine cations (HDPA) exhibit orientation disorder, and one of the water molecules exhibits positional disorder.
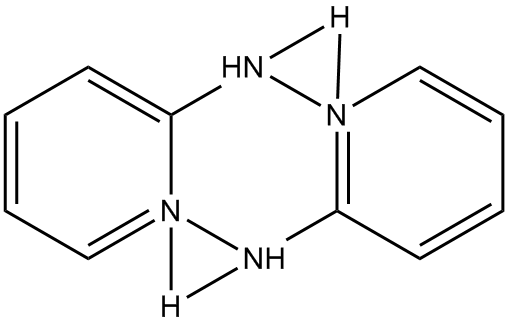
In EMETAS, HDPA exists as the cation. Because of symmetry disorder, the structure reported in EMETAS is a superposition of the various possible geometries. Even a cursory inspection of the reported structure, see Fig. 1, shows that there are severe problems, for example, the ring nitrogen atoms are bonded to the amine nitrogen, in variance with the known structure of HDPA. An idealized structure for this heterocycle is shown in Fig. 2.
|
Figure 1: Structure of
bis(2-(2-pyridyl)pyridinium) in EMETAS |
|
|
|
Figure 2: Idealized structure for bis(2-(2-pyridyl)pyridinium) |
|
|
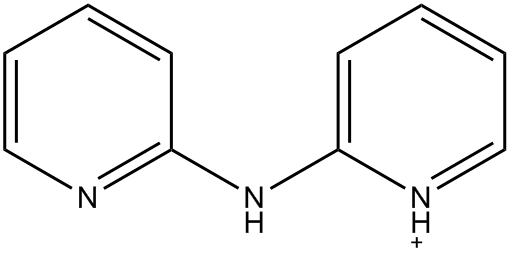
Before attempting to resolve this disorder, the relative energies of the various conformers were calculated. If the conformer shown is identified as "cis - cis" meaning that the two heterocycle nitrogen atoms are on the same side of the molecule, then the other conformers would be "cis - trans" and "trans - trans," "trans" meaning that the pyridyl group would be rotated by 180° about the C-NH bond, relative to the orientation in Figure 2. Both PM7 (Aq) and B3LYP/6-311G SCRF (DFT in solution) predict that the lowest-energy conformer would be "trans - trans", i.e., that the idealized geometry in Figure 2 was unlikely to be the correct conformation.
A MOPAC data-set was constructed for EMETAS. Because discrete species could not be isolated - the ATP complex would obviously interact electrostatically with the HDPA cations and with the magnesium(ii) hexahydrate - a solid-state system was used. Each disordered HDPA cation was then edited to form the trans - trans system.
Hydrogen atoms were added using the ADD-H utility in MOPAC.
One of the water molecules was disordered by symmetry. This gave rise to two equivalent water molecules, separated by a small distance; in EMETAS this was represented by two oxygen atoms that were close enough that they appeared to be bonded together. If this incorrect system was hydrogenated, it would give rise to hydrogen peroxide. Because each oxygen atom had a 50% occupancy, one of the oxygen atoms was deleted to make a chemically sensible system. The choice of which oxygen atom to delete was arbitrary and unimportant.
Steps in preparing
a MOPAC data-set for the EMETAS system
Conversion from CSD
to MOPAC format
Using MERCURY, a 2x2x2 multiple unit cell was constructed and saved in PDB format. Using MOPAC, this PDB file was converted into Cartesian coordinates, and the new file, in MOPAC data-set format, i.e., three blank lines then the Cartesian coordinates, saved as Make_EMETAS.mop.
Conversion to a
solid-state data-set or Large Unit Cell format
The three atoms that atom 1 would be translated to by the operations (1,0,0), (0,1,0), and (0,0,1) were identified. These atoms were re-named Tv to indicate that they were translation vector atoms.
Using MAKPOL, Make_EMETAS.mop was converted into a normal MOPAC data set for a solid-state calculation. The unit-cell in EMETAS was large enough that the default, MERS=(1,1,1), was suitable. The new data-set was automatically named EMETAS.mop.
Addition of
hydrogen atoms
EMETAS.mop was edited to add keyword ADD-H and run using MOPAC. This produced the hydrogenated system in which every site, except the magnesium atoms, was neutral. Hydrogen atoms were added and deleted to make the more realistic system. This involved adding a hydrogen atom to one or other of the heterocyclic nitrogen atoms in each of the eight DPA molecules (plus 8 H), deleting all hydroxyl hydrogen atoms on the phosphate groups on each of the eight ATP molecules (minus 32 H), and adding a hydrogen atom to ionize N1 on each of the 8 adenine groups (plus 8 H), for a net change in the number of hydrogen atom of -16. This resulted in a net change in charge of -16, which exactly balanced the +16 charge from the eight Mg2+ ions. Coordinates of all hydrogen atoms were then optimized to give the Opt-H structure.
Unconstrained
Geometry Optimization
The geometry of EMETAS was optimized using PM6. PM7 was not used because earlier work had shown that the PM7 geometry was not good. The optimized PM6 geometry is similar to the X-ray structure.
Interesting
features
Disorder in
orientation of the HDPA cation
Each HDPA cation can assume one of four possible positions. The amine group NH can be on one side or the other, and the heterocycle proton can be on one nitrogen or the other. Regardless of which position a specific HDPA cation has, it forms two strong hydrogen bonds. Two of the four HDPA cations each forms hydrogen bonds with oxygen atoms on PB of two ATP molecules, the other two HDPA cations each forms hydrogen bonds with two water molecules which, in turn, form hydrogen bonds to phosphate groups.
Ionization of
Adenine
When displayed in ConQuest, each adenine group in EMETAS is formally ionized with a proton being assigned to N1. In the solid state, that proton is near to an oxygen atom on PG of a different ATP molecule. When the positions of the hydrogen atoms was optimized, all other atoms being held fixed, the proton on each adenine migrated to the oxygen on a PG. This phosphate, unlike PA and PB, has a formal -2 charge. When the geometry was allowed to relax using PM6, the proton in seven out of the eight adenine groups moved back to the adenine, in the eighth group it stayed on the PG. So the question is, which is correct? An examination of related entries in the CSD does not provide an answer. Some adenine groups are assigned as ionized, some as neutral. In most instances the driving force can easily be identified, either a strong acid anion or a Group IA or IIA cation forced the adenine to be neutral or ionized. But in ATP, the ionization state of the triphosphate is not obvious. An alternative approach would be to look at the oxygen - nitrogen distance. If the PG phosphate was only singly ionized and the adenine was neutral, the O - N distance would be expected to be larger than if the PG phosphate was doubly ionized and the adenine was a cation. Examination of the PM6 structure showed that the O - N distance predicted using PM6 was the same as that in the CSD file, about 2.53 - 2.61 Ångstroms.
After this exercise was complete, the original journal article (Tamasia G, Berrettinib F, Hursthousec M, Cinia R. Effect of Free Water Molecules on the Structure of Mg-ATP-Dipyridylamine and Overview on Selected Metal-Adenosine Triphosphate Structures in Model Compounds and in Enzymes. Open Crystallography Journal. 2010;3:1-13) was examined, and in it the authors noted that "Bond distances and angles of phosphate group and purine moieties suggest that the acidic proton for the HATP3- ligand is disordered and resides both on a γ-phosphate oxygen atom and on N1 nitrogen atom from adenine. Thus, both O8 and N1 were considered protonated and the occupancies of both hydrogen atoms were fixed at 0.5." This provides a good example of the old adage that "A month in the laboratory can often save an hour in the library."
Comparison of CSD and PM6 geometries
Using the MOPAC "COMPARE" option, the geometry of the optimized PM6 structure was compared with the hydrogenated CSD Opt-H structure, both as output and graphically. As expected, the largest differences in bond-length involved atom pairs that were involved in the disorder of the HDPA cations. These differences ranged from 0.100 to 0.148 Ångstroms. The largest difference, 0.094 Ångstroms, attributable to errors in PM6 involved a P-O bond. On the other hand, PM6 accurately reproduces the six-membered Mg-O-P-O-P-O rings, and the slightly distorted octahedral [MgII(H2O)6]2+ ions.
Other features
The experimental density of EMETAS is reported as 1.534. The Opt-H structure had the same unit cell dimensions as the CSD EMETAS but had a density of 1.568. The difference was traced to the number of water molecules, seven per formula unit, in the calculated structure, whereas in the description of the CSD structure there were six water molecules. The optimized PM6 structure had a density of 1.618, or 3.2% higher than that of the Opt-H structure. Where the extra water molecule came from was, and is, an unsolved mystery.