 These missing bonds were added using keyword CVB, using the
multi-line keyword structure:
These missing bonds were added using keyword CVB, using the
multi-line keyword structure:A very common step in the preparation of systems to be modeled is the addition and removal of hydrogen atoms. This operation is both important and difficult. Important because computational models must be as realistic as possible, and difficult because errors in hydrogenation are inevitable, some due to faults in the starting geometry, and others due to the default hydrogenation procedure not accounting for certain specific conditions, particularly those that occur in large ring complexes. Data-sets can be edited to add or delete hydrogen atoms "by hand," and graphics utilities exist that allow data-sets to be edited easily. An alternative when working with MOPAC is to use keywords instead. This allows all the editing operations to a reference file, for example a PDB file from the Protein Data Bank, to be recorded in MOPAC data-sets, and if, as often happens, faults are found in the hydrogenation, these can easily be corrected by editing the appropriate data-set. Because some proteins are extremely complicated, a useful strategy is to use several data-sets to convert a PDB file into a realistic MOPAC data-set. The individual data-sets can be summarized as follows:
Template procedure for hydrogenating proteins
1. Using keyword ADD-H, hydrogenate the PDB file.
2. Using keyword CHARGES, identify all the ionized sites in the PDB file
from Step 1.
3. Using keyword SITE, correct the errors in hydrogenation in the PDB file
from Step 1.
4. Using keyword CHARGES, identify all the ionized sites in the PDB file
from Step 3.
5. Using keyword SITE=(SALT), add in all the default salt bridges.
6. Using keyword CHARGES, identify all the ionized sites in the PDB file
from Step 5.
7. Using keywords NOOPT and OPT-H, optimize the positions of the hydrogen
atoms in the PDB file from Step 5.
When faults are found in the ionized sites, the previous data-set should be edited to correct the faults, and re-run. Then the subsequent data-sets should be run. Eventually there should be no faults from hydrogenation.
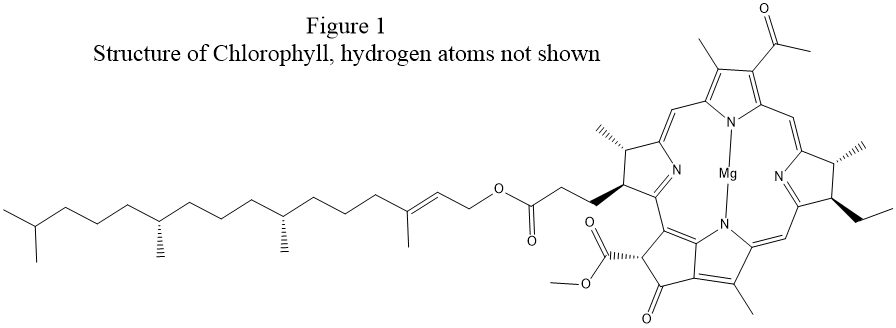
At this point, a valid question would be, why doesn't the program hydrogenate the PDB file correctly? There are several reasons. First, some PDB files have geometric errors. An example can be illustrated using bacteriochlorophyll. There are seven chlorophyll molecules in this protein, in one of these, BCL-3, a ketone C=O distance is given as 1.303 Å instead of the expected 1.22 Å. This distortion caused the ADD-H procedure to assign a hydrogen atom to the oxygen, making it a hydroxy group instead of a ketone. Mistakes of this type are easy to make, and need to be corrected. Second, in other systems where there are two possible hydrogenation sites, ADD-H sometimes picks the wrong atom. This occurs most often in histidine. Third, because most PDB files do not discriminate between them, the choice of which oxygen atom in a carboxylate group should be the hydroxy and which the keto is difficult to decide a priori. Given all this, it is easier to perform a simple hydrogenation then correct the errors in a subsequent job.
Worked example of the hydrogenation of Bacteriochlorophyll (PDB: 4BCL)
Bacteriochlorophyll is a large, complicated protein, and can be used to illustrate the various problems in hydrogenation, and the different steps in resolving them. All the files used can be found in Files used in Hydrogenation of Bacteriochlorophyll .zip.
Names of the data-sets used in this example
1 ADD-H.mop
2 Calculate charges in ADD-H structure (first attempt).mop
2 Calculate charges in ADD-H structure.mop
3 Modify ADD-H structure.mop
4 Re-calculate charges.mop
5 Add salt bridges.mop
6 Re-calculate charges after adding salt bridges.mop
7 PM6-D3H4 Optimize positions of hydrogen atoms.mop
7 PM7 Optimize positions of hydrogen atoms.mop
Step 1:
ADD-H: This is the only easy step. The entire keyword line
consists of:
GEO_DAT="Bacteriochlorophyll (4BCL).ent"
OUTPUT HTML ADD-H
NOCOMMENTS
The purpose of each keyword used here is as follows:
GEO_DAT="Bacteriochlorophyll (4BCL).ent": To minimize the
risk of making a mistake, it is preferable to have the geometry in a separate
file. So the original PDB file downloaded from the Protein Data Bank is
stored in the file
"Bacteriochlorophyll (4BCL).ent" By using the alternative suffix ".ent"
there is no danger that the original file could be accidentally modified by
MOPAC.
OUTPUT: In this and all subsequent operations there is no need to look
at the geometry in the ".out" file, so this minimizes the output, allowing only
material of interest to be printed.
HTML: Errors in hydrogenation are most easily found using a GUI.
HTML generates a simple web-page that can be read using FireFox and
JSmol also generates a PDB file. This file will then be used in the next
step, see below.
NOCOMMENTS: In addition to the geometry the original PDB file contains
a large amount of material that's not wanted here. None of that material is
relevant to the work being described here, so all of it can be ignored from this
point on.
Step 2: CHARGES: A first attempt to identify the charges resulting
from Step 1 using keywords GEO_DAT="1 ADD-H.pdb" OUTPUT CHARGES HTML
resulted in a large number of spurious errors caused by missing covalent
bonds. These errors were caused by the way metal atoms are treated in MOPAC; by
default, for the purpose of generating Lewis structures, magnesium atoms are
considered to be 100% ionic, i.e., not forming any covalent bonds. But an
examination of the structure of chlorophyll, see Fig. 1, shows that a
more realistic Lewis structure could be made by adding covalent bonds between
each magnesium atom and two nitrogen atoms on opposite sides of the ring.
These missing bonds were added using keyword CVB, using the
multi-line keyword structure:
GEO_DAT="1 ADD-H.pdb" OUTPUT CHARGES HTML ++
CVB=(++
"[BCL]1:B.MG":"[BCL]1:B.NB","[BCL]1:B.MG":"[BCL]1:B.ND",++
"[BCL]2:B.MG":"[BCL]2:B.NB","[BCL]2:B.MG":"[BCL]2:B.ND",++
"[BCL]3:B.MG":"[BCL]3:B.NB","[BCL]3:B.MG":"[BCL]3:B.ND",++
"[BCL]4:B.MG":"[BCL]4:B.NB","[BCL]4:B.MG":"[BCL]4:B.ND",++
"[BCL]5:B.MG":"[BCL]5:B.NB","[BCL]5:B.MG":"[BCL]5:B.ND",++
"[BCL]6:B.MG":"[BCL]6:B.NB","[BCL]6:B.MG":"[BCL]6:B.ND",++
"[BCL]7:B.MG":"[BCL]7:B.NB","[BCL]7:B.MG":"[BCL]7:B.ND"++
)
For convenience, atoms were identified by their Jmol name, not by their PDB name. Also, for ease of checking the CVB keyword for faults, each of the sets of Mg-N bonds was put on its own line.
Step 3: SITE: When the geometry in Step 2 was run the output showed that three cationic sites were predicted to exist. Because the purpose of the ADD-H operation is to add hydrogen atoms to satisfy valence requirements, if no errors occur then, with the exception of metal and isolated halogen atoms, all atoms should be neutral. So except for metals and halogen atoms the presence of ionized atoms normally indicates an error in hydrogenation. The ionized atoms are, in PDB notation:
HETATM 5564 C3B BCL 6
HETATM 5714 C4C BCL 4
HETATM 5864 C2D BCL 7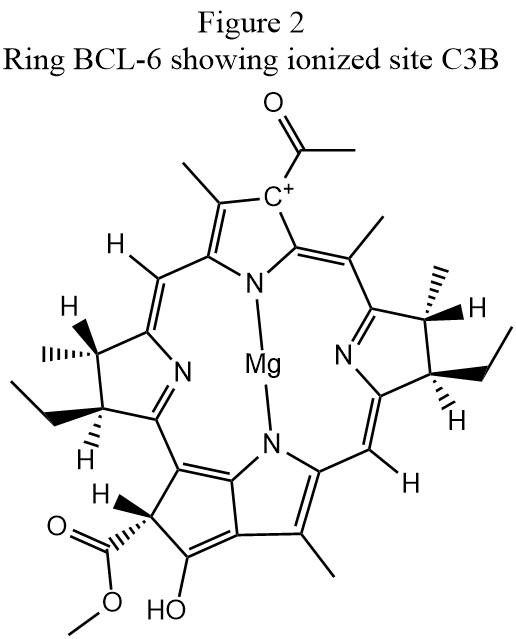
Identifying the cause of these faults is often quite difficult. In the case of the first ionized site, "HETATM 5564 C3B BCL 6", the atom ionized is indicated at the top of the ring in Fig. 2 as "C+" A superficial examination of the conjugated π-ring structure would suggest that this atom should either by hydrogenated or that there is a fault in the conjugation, but a careful comparison of the hydrogenation in Figs. 1 and 2 shows that the keto group an the bottom of Fig. 1 was incorrectly hydrogenated in Fig. 2. If the spurious hydrogen atom in the hydroxyl at the bottom of Fig. 2 is removed, the conjugation changes to be the same as that in Fig. 1 and carbon atom C3B is neutral, as expected.
Similarly, because of incorrect hydrogenation of atom C4A in BCL-4, the ring conjugation was incorrectly constructed, and "HETATM 5714 C4C BCL 4" was predicted to be ionized. When the hydrogen atom on C4A was removed, the correct conjugation formed, and C4C was correctly predicted to be neutral.
Incorrect hydrogenation of atom C1C in BCL-7 resulted in ionization of the third ion, "HETATM 5864 C2D BCL 7". As with the other two ions, correct conjugation was achieved by deleting the incorrect hydrogen atom.
A final examination of BCL-7 showed that a hydrogen atom had been incorrectly assigned to CHA and C1A. These and the other faulty hydrogen atoms were removed using the SITE command using the keywords:
GEO_DAT="2 Calculate charges in ADD-H structure.pdb" OUTPUT HTML ++Step 4: Re-calculate CHARGES: The results of Step 3 were checked in the same way as Step 2 was used to check the results of Step 1. The only difference being that instead of:
GEO_DAT="1 ADD-H.pdb"
in Step 2, the keyword in Step 4 was changed to:GEO_DAT="3 Modify ADD-H structure.pdb"
Step 5: SITE=(SALT): Although salt bridges could be added in Step 1, errors in ionization could be more easily identified by delaying adding salt bridges until after all ionization errors were corrected. They are added at this point using the keywords:
GEO_DAT="4 Re-calculate charges.pdb" OUTPUT HTML SITE=(SALT)
On running this job 17 salt bridges are added.
Step 6: Re-calculate CHARGES: This operation is similar to those in Steps 2 and 4. No errors were found, so the addition of hydrogen atoms could be considered to be correctly done.
Step 7: OPT-H: Positions of all the hydrogen atoms are optimized using PM7 and using PM6-D3H4. The complete set of keywords used for the PM7 optimization is as follows:
GEO_DAT="6 Re-calculate charges after adding salt bridges.pdb"
OUTPUT HTML CHARGE=0 NOOPT OPT-H MOZYME EPS=78.4 LET(100) ++
CVB=(++
"[BCL]1:B.MG":"[BCL]1:B.NB","[BCL]1:B.MG":"[BCL]1:B.ND",++
"[BCL]2:B.MG":"[BCL]2:B.NB","[BCL]2:B.MG":"[BCL]2:B.ND",++
"[BCL]3:B.MG":"[BCL]3:B.NB","[BCL]3:B.MG":"[BCL]3:B.ND",++
"[BCL]4:B.MG":"[BCL]4:B.NB","[BCL]4:B.MG":"[BCL]4:B.ND",++
"[BCL]5:B.MG":"[BCL]5:B.NB","[BCL]5:B.MG":"[BCL]5:B.ND",++
"[BCL]6:B.MG":"[BCL]6:B.NB","[BCL]6:B.MG":"[BCL]6:B.ND",++
"[BCL]7:B.MG":"[BCL]7:B.NB","[BCL]7:B.MG":"[BCL]7:B.ND"++
)
NOOPT is used because the input geometry is in PDB format, and,
since that format does not include optimization flags, when a geometry in PDB
format is read in all optimization flags are turned on by default.
OPT-H: In this job, OPT-H indicates that the positions of all
hydrogen atoms should be optimized.
MOZYME: Essential for large systems.
EPS=78.4: The COSMO implicit-solvation model to be used.
LET(100): Higher accuracy in geometry optimization that the default is used.
For the PM6-D3H4 optimization, keyword PM6-D3H4 is
added.
Examples where ADD-H produces systems that have ions
In 5UVJ four ions, a [Na]+ and three [Cl]-, are present. These are ionized by default, resulting in a net charge of -2. This is not a fault.
In 5VPS there is a heterogroup, 9G1, that contains a phosphanium (phosphonium). In 9G1, phosphorus forms single bonds with carbon atoms on four ligands. This results in the heterogroup having a net charge of +1, and thus the entire system has a net charge of +1. This, also, is not a fault.
The PDB geometry of 5VPS does have a problem with 9G1, in that the C-C and C-N distances in the piperidine ring, click on "Specific Script" to see this, are very short, averaging 1.36 Å instead of the expected 1.51 Å. Both the topology and the bond-lengths of 9G1 in 5VPS suggest that the hexagonal C5N ring should be a 4-(H)-pyridinium rather than a piperidine. This error in hydrogenation affects only C2, C3, C5, and C6, and therefore does not introduce an error in the net charge, but should obviously be corrected by adding hydrogen atoms. Errors of this type can also be corrected by use of the SITE keyword.
In 5ULI there is a heterogroup, 0WD, that appears to contain a pyridinium ion - both C3-C4 and C4-C5 distances in the hexagonal C5N ring in 5ULI are about 1.40 Å. This is nearer to the expected equivalent distances in pyridine, 1.37 Å, than to that in a 4-(H)-pyridine. In 0WD these distances should be about 1.49 - 1.50 Å. Both the topology and the bond-lengths of 0WD in 5ULI incorrectly suggest that the hexagonal C5N ring should be a pyridinium rather than a 1,4-dihydropyridine. This fault results in the ring having a net charge of +1. This type of error can only be detected by a careful examination of the published structure. Once detected, correcting faults of this type by the use of keyword SITE is straightforward.
|
Detail of the 1,4-dihydropyridine ring in 0WD in 5ULI | |
| PDB Geometry | PM7 Geometry |
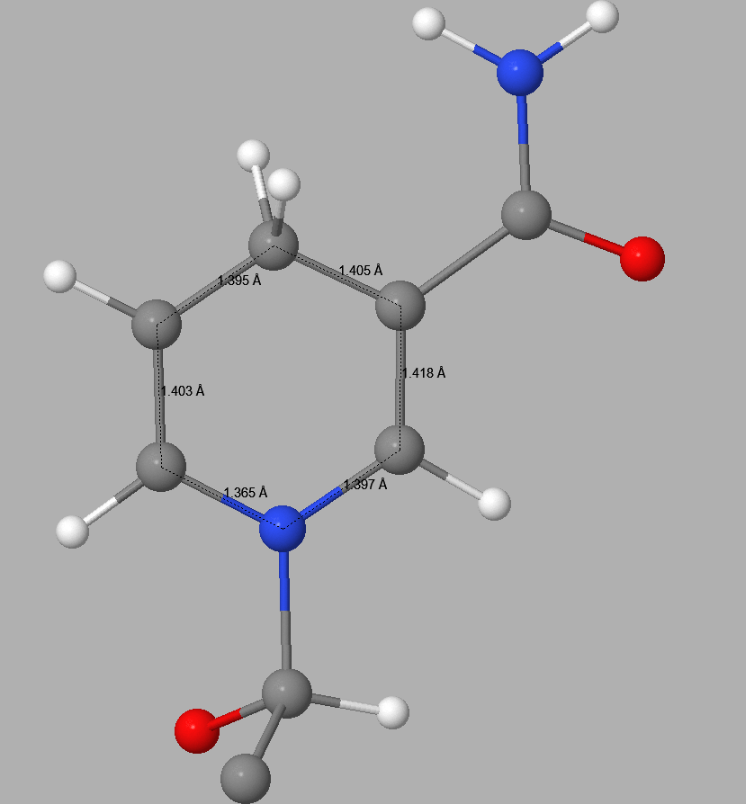 | 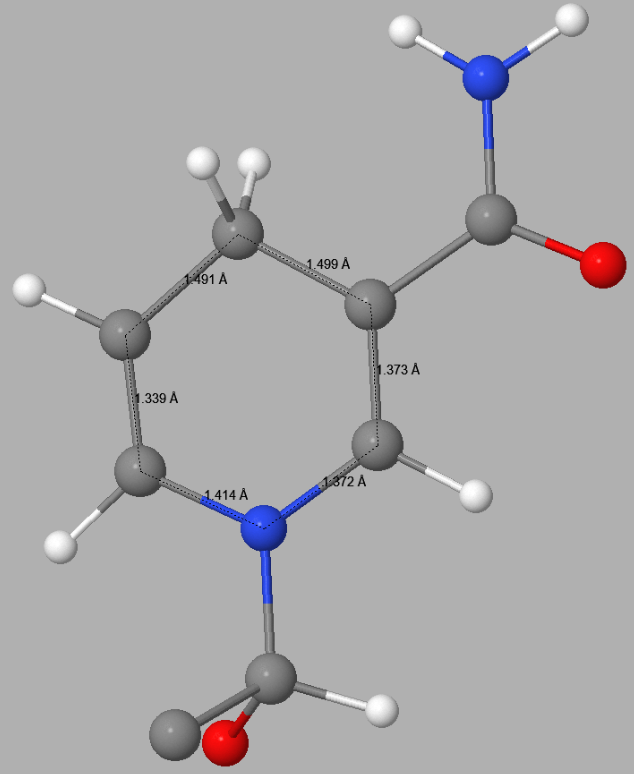 |
|
To see this in the protein click on "Specific Script" |
To see this in the protein click on "Specific Script" |
In general, hydrogenation using ADD-H is done correctly. No errors were detected in validation tests for a set of proteins