For most systems, a calculation run using the linear scaling method MOZYME will produce the same result as a calculation run using conventional SCF methods. In some systems, however, a MOZYME calculation will result in a heat of formation that is significantly greater than that predicted using conventional methods. The questions that come to mind are:
(A) Under what conditions would MOZYME produce a DHf
that is significantly greater than that predicted by conventional methods?
(C) What is the cause of the DHf
being different, depending on how the SCF equations are solved?
(D) What are the implications of the different DHf?
| DHf (Kcal/mol) Gas-phase | |||
| n | MOPAC | MOZYME | Diff |
| 1 | -31.45 | -31.45 | 0.00 |
| 2 | 2.10 | 2.10 | 0.00 |
| 3 | 18.31 | 18.32 | 0.01 |
| 4 | 33.23 | 37.15 | 3.92 | 5 | 41.14 | 49.92 | 8.78 |
| 6 | 44.39 | 57.89 | 13.50 |
| 7 | 43.63 | 58.81 | 15.18 |
| 8 | 46.44 | 69.29 | 22.85 |
| 9 | 48.75 | 73.76 | 25.01 |
| 10 | 42.39 | 69.03 | 26.64 |
The MOZYME technique starts with a Lewis structure. Lewis structures for
most systems are straightforward, thus in a carboxylic acid group, -COOH, the
oxygen atom that is singly bonded to the carbon atom is also singly bonded to
the hydrogen atom, and has two lone pairs. If the hydrogen atom is removed, then the
oxygen atom it was attached to is given three lone pairs, and a negative charge,
resulting in a -COO(-), or carboxylate group. The resulting electric charge could be
stabilized by a nearby positive charge, and would be de-stabilized by a nearby
negative charge.
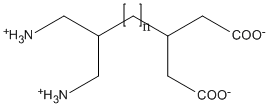
Consider now the systems consisting of a number, n, of methylene groups forming a chain, at one end there is a doubly-ionized glutarate group, and at the other end is a doubly-ionized 1,3-diammino propyl group, see figure. Single SCF calculations (1SCF) were run on each system using the conventional SCF calculation, call this MOPAC, and also using the MOZYME method of solving the SCF equations. For small values of n, MOPAC gave the same result as a MOZYME calculation, see table headed "Gas-phase," but for larger values of n, the MOZYME results gave DHf that were greater than the MOPAC results. Unless a mistake is made, by definition, the lower the energy the better, so there is an apparent fault in the MOZYME results.
An examination of the charges on the atoms in the systems where the MOPAC and MOZYME results are different shows that in the MOPAC results the net charge on the diammine end converges on +1.0 as n increases, while the MOZYME charge stays at ca. +2.0. A similar trend occurs at the di-carboxylate end, where the MOPAC charge converges on -1.0 and the MOZYME charge stays at ca. -2.0. The effect of the charged ends is also reflected in the dipole moments. For n=10, these are 97.5D (MOPAC) and 151.7D (MOZYME).
The systems being used are, however, very artificial. In the MOPAC calculations, the energy can be lowered by forming a radical, where two of the electrons on the di-carboxylate end are moved to the diammine end of the molecule. That this occurs can be verified by running a UHF calculation on the various systems. Where the MOPAC and MOZYME results agree, the <S2> is zero, where they differ, the <S2> is non-zero.
| DHf (Kcal/mol) In water | |||
| n | MOPAC | MOZYME | Diff |
| 1 | -224.74 | -224.73 | 0.01 |
| 2 | -232.52 | -232.51 | 0.01 |
| 3 | -236.79 | -236.78 | 0.01 |
| 4 | -235.95 | -235.95 | 0.00 | 5 | -240.18 | -240.17 | 0.01 |
| 6 | -244.97 | -244.96 | 0.00 |
| 7 | -255.19 | -255.19 | 0.00 |
| 8 | -254.27 | -254.27 | 0.00 |
| 9 | -259.07 | -259.06 | 0.01 |
| 10 | -268.99 | -268.99 | 0.00 |
Does this represent a fault or limitation of the MOZYME method? In short, the answer is no, for several reasons:
Archive files, that can be used as MOPAC data-sets, for MOPAC and MOZYME calculations in gas-phase and in solution can be downloaded.